Welcome to use iKa/Ks
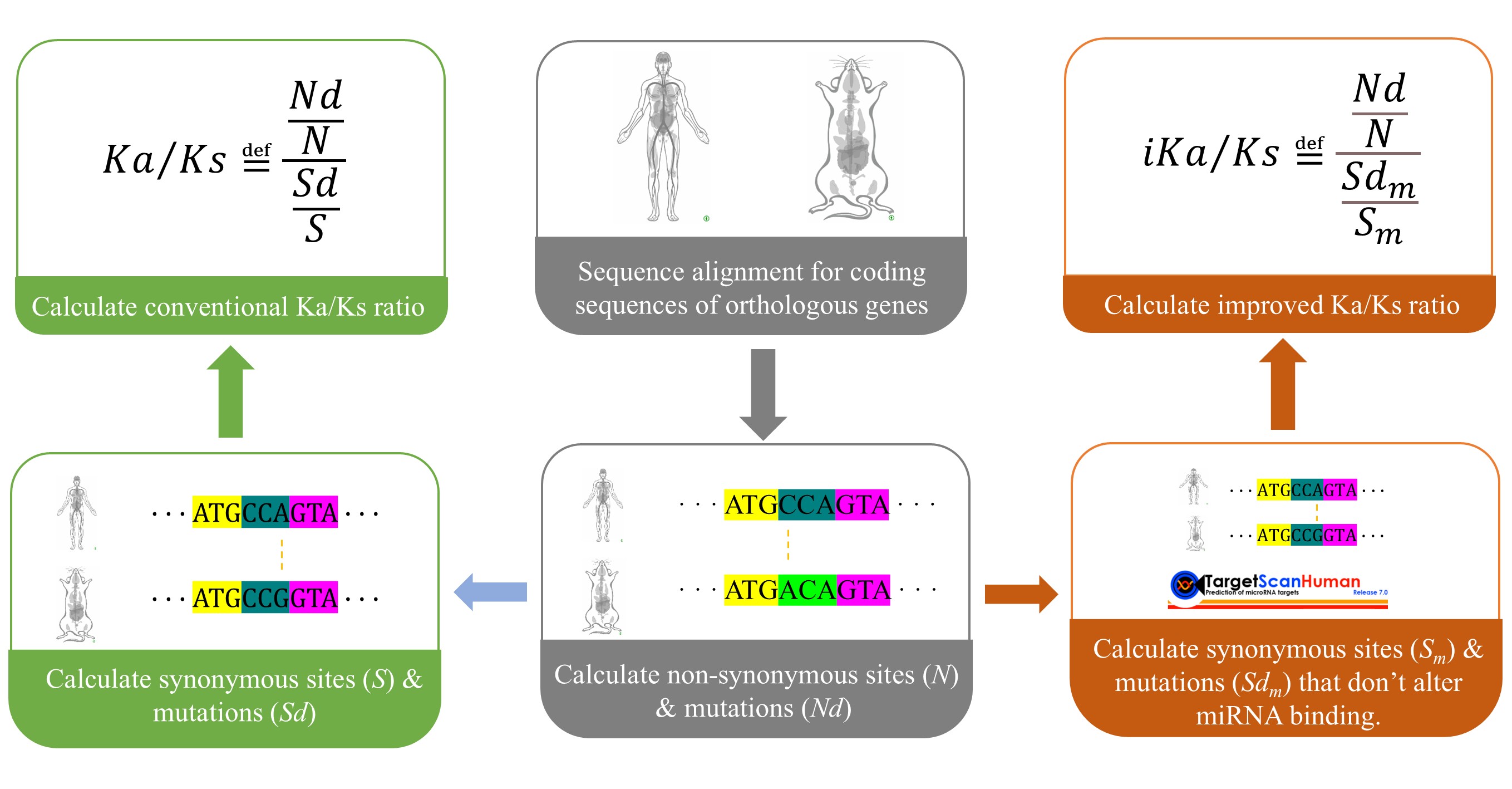
iKa/Ks is a novel algorithm to estimate the non-synonymous/Synonymous substitution rate (Ka/Ks) ratio, a commonly used metric to estimate the selection pressure and evolutionary rate of proteins in comparative genomics, which plays critical roles in molecular evolution in both biology and medicine. The core contribution of iKa/Ks is that it breaks down the fundamental assumption of Ka/Ks, synonymous mutations are evolutionarily neutral and not subject to natural selection, by introducing the altered status of miRNA regulation of the synonymous mutations. (Ye et al. J Mol Biol. 2026).
Currently, users can download:
Data Collection
We obtained the mature coding sequences of all orthologous genes between human (Homo sapiens, GRCh38) and mouse (Mus musculus, GRCm39) from Ensembl, prioritizing the longest transcript sequence for genes with multiple mature transcripts. To ensure accurate Ka/Ks calculation, we performed codon-aware sequence alignment using ParaAT (Parallel Alignment and back-Translation), a tool designed specifically for this purpose. ParaAT first aligns protein sequences using MAFFT (default parameters), then performs back-translation to generate codon-aware nucleotide alignments that respect codon boundaries. Sites containing gaps were removed prior to downstream analysis. This procedure yielded a total of 22,130 high-quality orthologous gene pairs for subsequent analysis.
Contact Us
Dr Qinghua Cui, 38 Xueyuan Rd, Department of Biomedical Informatics, Peking University Health Science Center, Beijing 100191, China
Email: cuiqinghua@bjmu.edu.cn
Citing iKaKs
Ye, Jiachen et al. “iKa/Ks: Estimating the Selection Pressure and Evolutionary Rate of Proteins under the Non-neutral Hypothesis of Synonymous Mutations.” Journal of molecular biology, 169895. 9 Jun. 2026, doi:10.1016/j.jmb.2026.169895