Tutorial
Here you can find answers to common questions and instructions on how to use microAF.
📚 What is microAF?
microAF is a comprehensive human microprotein structure database that integrates large-scale smORF-derived protein data with high-precision structural predictions. The current release includes 617,462 human smORF nucleotide sequences and their corresponding microprotein translations.
Among them, over 370,000 microprotein structures have been accurately predicted using AlphaFold2, with ongoing updates to expand coverage. The predicted structures have an average PLDDT score of 71.95, indicating generally high confidence.
microAF also provides variant effect prediction using residue probability matrices derived from the ESM-2 650M language model, and subcellular localization prediction powered by the LocPro model.
Additionally, microAF integrates the Foldseek algorithm, enabling users to upload their own protein structure files (.pdb or .cif) and perform fast structural alignment against proteins in microAF and external resources such as the AlphaFold Protein Structure Database (AFDB).
Overall, microAF serves as a functional and structural resource for exploring the biological significance of microproteins at the proteome scale.
🧬 What is a microprotein?
Microproteins are a class of proteins typically composed of fewer than 100 amino acids, frequently encoded by small open reading frames (smORFs). Despite their diminutive size, microproteins have been increasingly recognized for their functional significance in various biological processes, including cellular signaling, gene regulation, and disease pathogenesis.
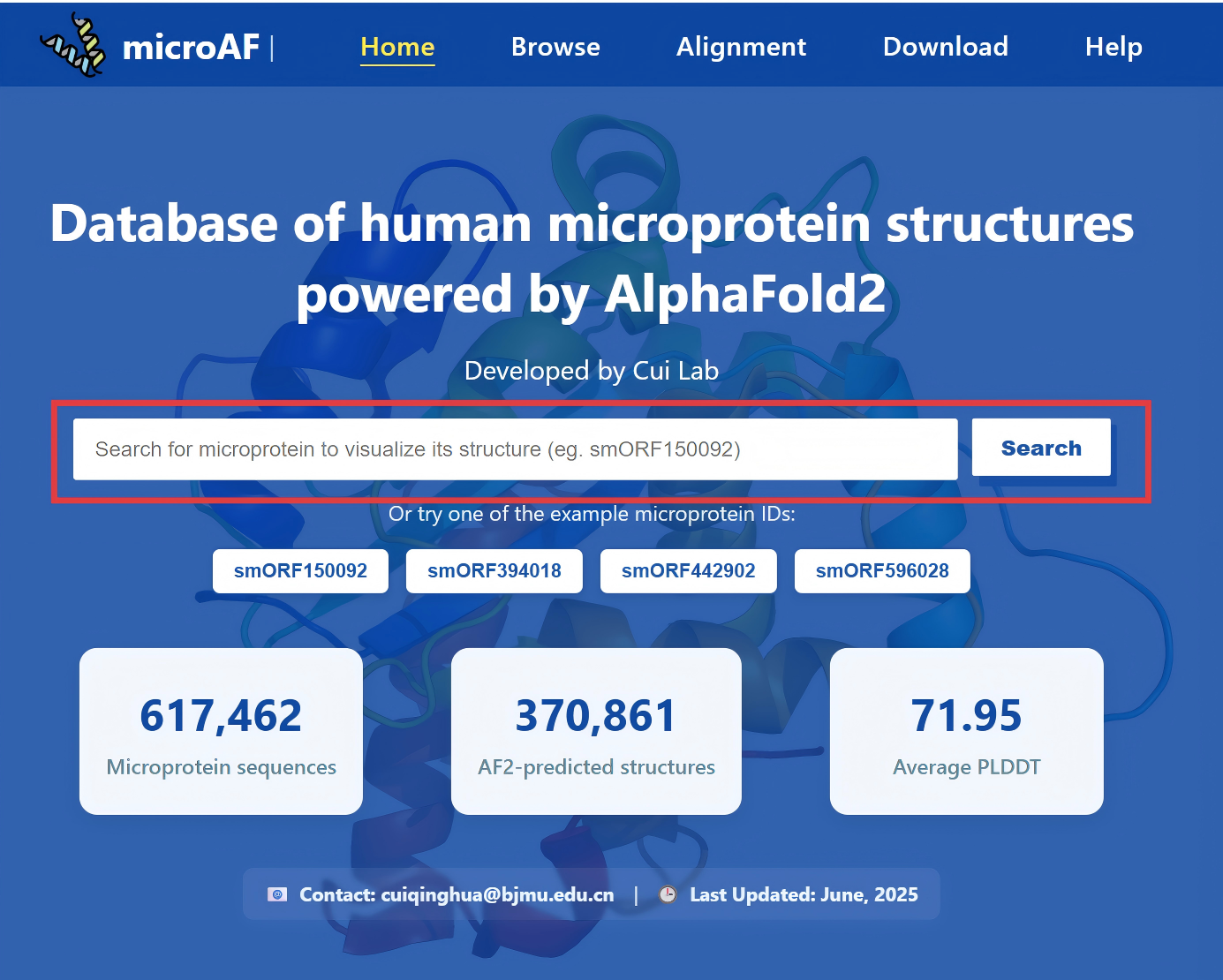
🔍 How do I search for a microprotein?
Go to the Home page and enter a smORF ID like smORF150092 into the search bar. Click "Search" to view its predicted structure and metadata.
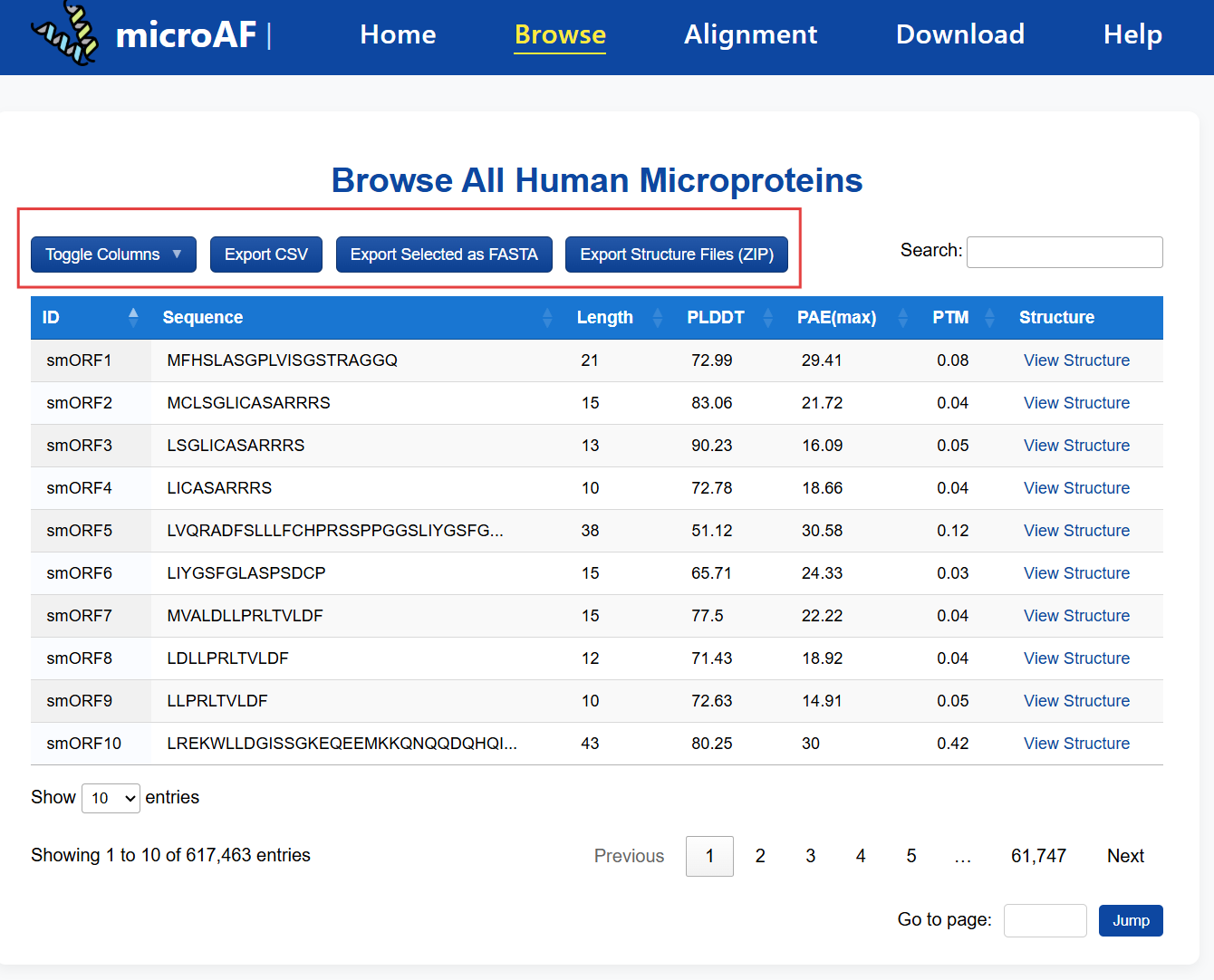
📥 How do I download sequences or structures of microproteins?
In the Browse page, select desired rows, then use the buttons above the table to export FASTA or download structure files in ZIP format.
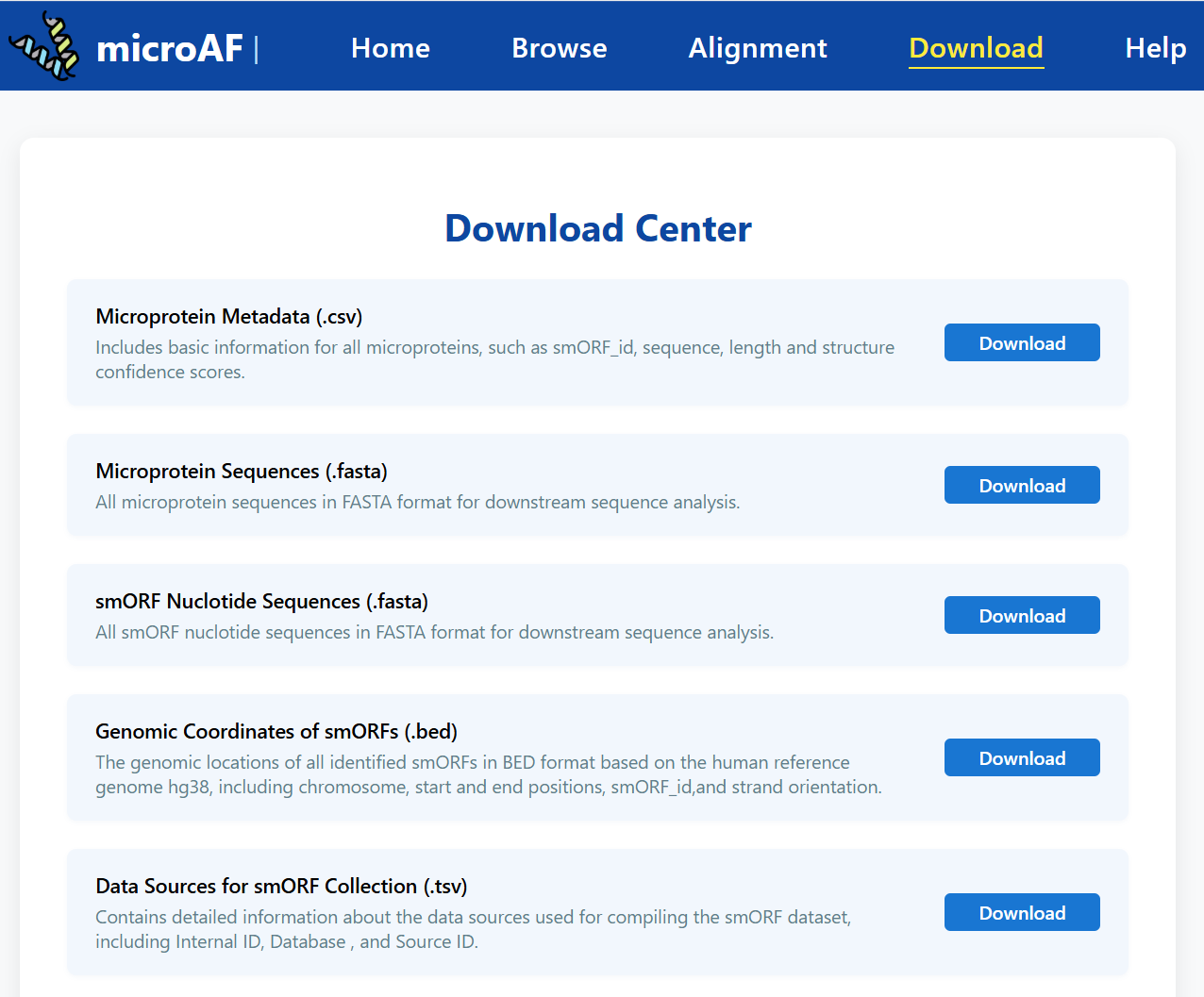
📦 How can I download the complete microprotein dataset?
To access the full set of microprotein sequences and structure predictions, please visit the
Download page.
There, you can download comprehensive files in CSV, FASTA, or structure (PDB/ZIP) formats.
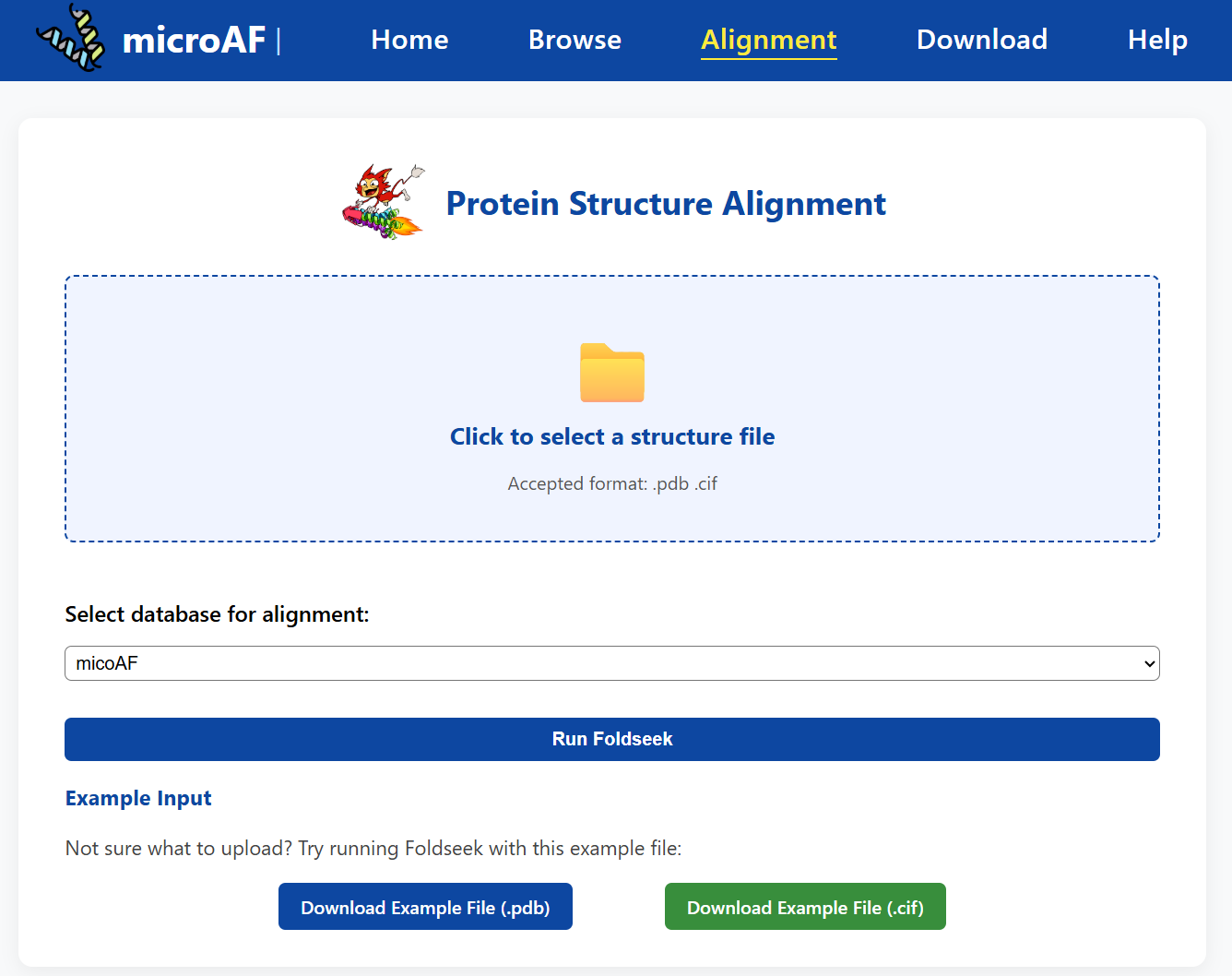
🔗 What is the purpose of the Alignment page?
The Alignment page enables users to perform structural comparisons between a query protein structure and reference databases using the Foldseek algorithm. By uploading a protein structure file in .pdb or .cif format, users can identify proteins with highly similar 3D conformations from datasets such as microAF, AlphaFold Protein Structure Database (AFDB), and others. This functionality is particularly useful for discovering potential structural homologs, inferring function from structural similarity, or validating predictions from novel or uncharacterized microproteins.
🧠 What do "Variant Effect Prediction" and "Subcellular Localization" mean?
The Variant Effect Prediction module estimates the functional impact of amino acid substitutions based on a per-residue log-likelihood matrix derived from the ESM-2 650M protein language model. This model provides a probability distribution over all possible amino acids at each position, allowing for in silico evaluation of mutation effects based on changes in sequence plausibility.
The Subcellular Localization Prediction module is powered by the LocPro model, which uniques in (a) combining protein representations from the ESM2 and the expert tool PROFEAT, (b) implementing a hybrid deep neural network with CNN, FC, and BiLSTM blocks, and (c) developing a multi-label framework for predicting protein subcellular localization at various granularities. Predictions span categories such as nucleus, cytoplasm, mitochondrion, and plasma membrane, among others.
Together, these tools provide insights into both the functional tolerance of sequence variants and the potential biological context in which the microprotein may act.
🧪 What is smORFunction?
smORFunction is a computational tool developed by our research group to predict the potential biological functions of microproteins.Functional predictions generated by smORFunction can assist in hypothesis generation and downstream experimental validation of novel microproteins.

🔗 Learn more: smORFunction Web Interface
🧬 How can I run custom bioactivity prediction?
HMPA now provides an interactive prediction page where you can submit your own microprotein sequence or upload a structure file (.pdb/.cif) and obtain bioactivity results in real time.
Open: Bioactivity Predictor
❓ What do the PLDDT, PTM, and PAE scores represent?
PLDDT (Predicted Local Distance Difference Test) is a per-residue confidence score ranging from 0 to 100 provided by AlphaFold2. It estimates the accuracy of the predicted position of each residue. Scores above 70 are typically considered reliable, while scores below 50 indicate low confidence.
PTM (Predicted TM-score) is a global measure of prediction confidence for the overall structure. It estimates the similarity of the predicted structure to the true structure on a scale from 0 to 1, with values above 0.7 generally suggesting high-confidence global topology.
PAE (Predicted Aligned Error) is a pairwise matrix indicating the expected positional error in angstroms (Å) between any two residues after optimal alignment. Lower PAE values imply higher certainty in the relative spatial positioning of residues or domains. PAE is particularly useful for assessing domain packing confidence.